Good Calories Bad calories
Good Calories Bad calories by tIM fERRIS
Last week, I had a wonderful conversation with Gary Taubes,
Good Calories bad calories, my favorite science journalist and author of the incredible (and I consider definitive), Good Calories, Bad Calories. His ability to synthesize and recall research, both in writing and in speaking, is one of the most amazing feats I’ve ever witnessed.
It is with great pleasure, therefore, that I offer you the director’s-cut chapter that didn’t make it into the book.
The chapter addresses important misconceptions about diet, fructose, blood pressure, and diabetes through the lens of gout.
If you don’t know someone with gout, you probably will. It is common and becoming more so. The misguided prescriptions from misinformed doctors, which Taubes addresses, have affected my family, and I’d rather save you the trouble if I can.
Good Calories Bad Calories
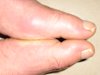
What the hell is gout anyway.
Like many, I’d heard it a million times but never knew. Here it is…
British physician Alfred Garrod, in the mid-19th century, identified uric acid as the causative agent; the idea being that uric acid accumulates in the circulation [and] crystallizes into needle-sharp urate crystals. These crystals then lodge in the soft tissues and in the joints of the extremities –- classically, the big toe — and cause inflammation, swelling and an excruciating pain that was described memorably by the 18th century bon vivant Sydney Smith as like walking on one’s eyeballs.
Sound like something to avoid?
Disclaimer as requested by Gary: This chapter is in draft form and has not gone through the same fact-checking as the rest of Taubes’ published work, even though there are 32 citations (some incomplete). I wanted to show the writing process at its mid-point. The only deletions I’ve make are “TK”, which–for unknown reasons–is traditional shorthand in publishing for indicating that something is “to come”.
I have bolded several sections for those who would like a 2-3-minute skim of content highlights before digesting the entire piece, which is 7 pages long.
Enter Gary Taubes:
—-
Gout and the condition known technically as hyperuricemia, or elevated levels of uric acid, are the most recent examples of this kind of institutional neglect of the potential health effects of fructose, and how pervasive it can be.
Gout itself is an interesting example because it is a disease that has gone out of fashion in the last century and yet the latest reports suggest it is not only as prevalent as ever, but becoming more so. Recent surveys suggest that nearly 6 percent of all American men in their fifties suffer from gout, and over ten percent in their seventies. The proportion of women afflicted is considerably less at younger ages but still rises over 3 percent by age 60.(1) Moreover, the prevalence of gout seems to have doubled over the last quarter century, coincident (perhaps not coincidentally) with the reported increase in obesity, and it may have increased five- or even six-fold since the 1950s, although a large portion of that increase may be due to the aging of the population.(2)
Until the late 17th century, when the spread of gout reached almost epidemic proportions in Britain, the disease afflicted almost exclusively the nobility, the rich and the educated, and so those who could afford to indulge an excessive appetite for food and alcohol. This made gout the original example of a disease linked to diet and over-consumption, and so, in effect, the original disease of civilization.
But once gout became easily treatable, in the early 1960s, with the discovery of the drug allopuranol, clinical investigators and researchers began to lose interest. And the pathology of gout has been understood since the British physician Alfred Garrod, in the mid-19th century, identified uric acid as the causative agent; the idea being that uric acid accumulates in the circulation to the point that it falls out of solution, as a chemist would put it, and so crystallizes into needle-sharp urate crystals. These crystals then lodge in the soft tissues and in the joints of the extremities – classically, the big toe — and cause inflammation, swelling and an excruciating pain that was described memorably by the 18th century bon vivant Sydney Smith as like walking on one’s eyeballs.(3) Because uric acid itself is a breakdown product of protein compounds known as purines – the building blocks of amino acids – and because purines are at their highest concentration in meat, it has been assumed for the past 130-odd years that the primary dietary means of elevating uric acid levels in the blood, and so causing first hyperuricemia and then gout, is an excess of meat consumption.
The actual evidence, however, has always been less-than-compelling: Just as low cholesterol diets have only a trivial effect on serum cholesterol levels, for instance, and low-salt diets have a clinically insignificant effect on blood pressure, low-purine diets have a negligible effect on uric acid levels. A nearly vegetarian diet, for instance, is likely to drop serum uric acid levels by 10 to 15% percent compared to a typical American diet, but that’s rarely sufficient to return high uric acid levels to normality, and there is little evidence that such diets reliably reduce the incidence of gouty attacks in those afflicted.(4) Thus, purine-free diets are no longer prescribed for the treatment of gout, as the gout specialist Irving Fox noted in 1984, “because of their ineffectiveness” and their “minor influence” on uric acid levels.(5) Moreover, the incident of gout in vegetarians, or mostly vegetarians, has always been significant and “much higher than is generally assumed.” (One mid-century estimate, for instance, put the incidence of gout in India among “largely vegetarians and teetotalers” at 7%.)(6) Finally, there’s the repeated observation that eating more protein increases the excretion of uric acid from the kidney and, by doing so, decreases the level of uric acid in the blood.(7) This implies that the meat-gout hypothesis is at best debatable; the high protein content of meats should be beneficial, even if the purines are not.
The alternative hypothesis is suggested by the association between gout and the entire spectrum of diseases of civilization, and between hyperuricemia and the metabolic abnormalities of Syndrome X. In the past century, gout has manifested all of the now-familiar patterns, chronologically and geographically, of diseases of civilization, and so those diseases associated with western diets. European physicians in World War I, for instance, reported a reduced incidence of gout in countries undergoing food shortages.(8) In primitive populations eating traditional diets, gout was virtually unknown or at least went virtually unreported (with the conspicuous exception of Albert Schweitzer who says he saw it with surprising frequency.) The earliest documented cases reported in Asia and Africa were in the late 1940s.(9) And even in the 1960s, hospital records from Kenya and Uganda suggested an incidence of gout lower than one in a thousand among the native Africans. Nonetheless, by the late 1970s, uric acid levels in Africa were increasing with westernization and urbanization,(10) while the incidence of both hyperuricemia and gout among South Pacific islanders was reportedly sky-rocketing. By 1975, the New Zealand rheumatologist B.S. Rose, a colleague of Ian Prior’s, was describing the native populations of the South Pacific as “one large gouty family.”(11)
Gout has also been linked to obesity since the Hippocratic era, and this association is the origin of the assumption that high-living and excessive appetites are the cause. Gouty men have long been reported to suffer higher rates of atherosclerosis and hypertension, while stroke and coronary heart disease are common causes of death.(12) Diabetes is also commonly associated with gout. In 1951, Menard Gertler, working with Paul Dudley White’s Coronary Research Project at Harvard, reported that serum uric acid levels rose with weight, and that men who suffered heart attacks were four times as likely to be hyperuricemic as healthy controls.(13) This led to a series of studies in the 1960s, as clinical investigators first linked hyperuricemia to glucose intolerance and high triglycerides, and then later to high insulin levels and insulin resistance.(14) By the 1990s, Gerald Reaven, among others, was reporting that insulin resistance and hyperinsulinemia raised uric acid levels, apparently by decreasing uric acid excretion by the kidney, just as they raised blood pressure by decreasing sodium excretion. “It appears that modulation of serum uric concentration by insulin resistance is exerted at the level of the kidney,” Reaven wrote, “the more insulin-resistant an individual, the higher the serum uric acid concentration.” (15)
These observations would suggest that anything that raised insulin levels would in turn raise uric acid levels and might cause gout, which would implicate any high carbohydrate diet with sufficient calories. But this neglects the unique contribution of fructose. The evidence arguing for sugar or fructose as the primary cause of gout is two-fold. First, the distribution of gout in western populations has paralleled the availability of sugar for centuries, and not all refined carbohydrates in this case. It was in the mid-17th century, that gout went from being exclusively a disease of the rich and the nobility to spread downward and outward through British society, reaching near epidemic proportions by the 18th century. Historians refer to this as the “gout wave,”(16) and it coincides precisely with the birth and explosive growth of the British sugar industry(17) and the transformation of sugar, in the words of the anthropologist Sydney Mintz, from “a luxury of kings into the kingly luxury of commoners.”(18) British per capita sugar consumption in the 17th century was remarkably low by modern standards, a few pounds per capita per year at the turn of the century, but the change in consumption over the next century and a half was unprecedented: between 1650 and 1800, following the British acquisition of Barbados, Jamaica and other “sugar islands”, total sugar consumption in England and Wales increased 20- to 25-fold.(19)
The second piece of evidence is much less circumstantial: simply put, fructose increases serum levels of uric acid. The “striking increase” in uric acid levels with an infusion of fructose was first reported in the Lancet in the late 1960s by clinicians from Helsinki, Finland, who referred to it as fructose-induced hyperuricemia.(20) This was followed by a series of studies through the late 1980s confirming the existence of the effect and reporting on the variety of mechanisms by which it came about. Fructose, for instance, accelerates the breakdown of a molecule known as ATP, which is the primary source of energy for cellular reactions and is loaded with purines. (ATP stands for adenosine triphosphate; adenosine is a form of adenine, and adenine is a purine.) And so this in turn increases formation of uric acid. Alcohol apparently raises uric acid levels through the same mechanism, although beer also has purines in it.(21) Fructose also stimulates the synthesis of purines directly, and the metabolism of fructose leads to the production of lactic acid, which in turn reduces the excretion of uric acid by the kidney and so raises uric acid concentrations indirectly by that mechanism.(22)
These mechanistic explanations of how fructose raises uric acid levels were then supported by a genetic connection between fructose metabolism and gout itself. Gout often runs in families, so much so that those clinicians studying gout have always assumed the disease has a strong hereditary component. In 1990, Edwin Seegmiller, one of the few veteran gout researchers in the U.S., and the British geneticist George Radda, who would go onto become director of the Medical Research Counsel, reported that the explanation for this familial association seemed to be a very specific defect in the genes that regulate fructose metabolism. Thus, individuals who inherit this defect will have trouble metabolizing fructose and so will be born with a predisposition to gout. This suggested the possibility, Seegmiller and Radda concluded, that this defect in fructose metabolism was “a fairly common cause of gout.”(23)
As these observations appeared in the literature, the relevant investigators were reasonably clear about the implications: “since serum-uric-acid levels are critical in individuals with gout, fructose might deserve consideration in their diet,” noted the Helsinki clinicians in The Lancet in 1967, and so the chronic consequences of high-fructose diets on healthy individuals required further evaluation.(24) Gouty patients should avoid high-fructose or high-sucrose diets, explained Irving Fox in 1984, because “fructose can accelerate rates of uric acid synthesis as well as lead to increased triglyceride production.”(25) Although none of these investigators seemed willing to define what precisely constituted a high-fructose or a high-sucrose diet. Was it 50 pounds of sugar a year? 100 pounds? 150 pounds? 300 pounds? And would high-fructose diets induce gout in healthy individuals or would they only exacerbate the problem in those already afflicted? In 1993, the British biochemist Peter Mayes published an article on fructose metabolism in the American Journal of Clinical Nutrition that is now considered the seminal article in the field. (This was in the special issue of the AJCN dedicated to the health effects of fructose.) Mayes reviewed the literature and concluded that high-fructose diets in healthy individuals were indeed likely to cause hyperuricemia, and he implied that gout could be a result, as well, but the studies to address that possibility had simply never been done. “It is clear,” Mayes concluded, “that systematic investigations in humans are needed to ascertain the precise amounts, both of fructose consumption and of its concentration in the blood, at which deleterious effects such as hyperlipidemia and hyperuricemia occur.”(26) Add to this Reaven’s research reporting that high insulin levels and insulin resistance will increase uric acid levels, and it suggests, as Mayes had remarked about triglycerides, that sugar (sucrose) and high fructose corn syrup would constitute the worst of all carbohydrates when it comes to uric acid and gout. The fructose would increase uric acid production and decrease uric acid excretion, while the glucose, though its effect on insulin, would also decrease uric acid excretion. Thus, it would be reasonable to assume or at least to speculate that sugar is a likely cause of gout, and that the patterns of sugar consumption explain the appearance and distribution of the disease.
Maybe so, but this hypothesis has never been seriously considered. Those investigators interested in gout have focused almost exclusively on alcohol and meat consumption, in part because these have historical precedents and because the implication that gouty individuals and particularly obese gouty individuals shy away from meat and alcohol fit in well with the dietary prescriptions of the 1970s onward.
More than anything, however, this sugar/fructose hypothesis was ignored, once again, because of bad timing. With the discovery and clinical application of allopurinol in the 1960s, those clinical investigators whose laboratories were devoted to studying the mechanisms of gout and purine metabolism – James Wyngaarden’s, for instance, at Duke and Edwin Seegmiller’s at NIH – began focusing their efforts either on working out the nuances of allopurinol therapy, or to applying the new techniques of molecular biology to the genetics of gout and rare disorders of hyperuricemia or purine metabolism. Nutritional studies were simply not considered worthy of their time, if for no other reason than that allopuranol allowed gout suffers to eat or drink whatever they wanted. “We didn’t care so much whether some particular food might do something,” says William Kelley, who is a co-author with Wyngaarden of the 1976 textbook, Gout and Hyperuricemia and who started his career in Seegmiller’s lab at NIH. “We could take care of the disease.”(27)
This exodus, however, coincided with the emergence of research on fructose-induced hyperuricemia. By the 1980s, when the ability of fructose and sucrose consumption to raise uric acid levels in human subjects was demonstrated repeatedly, the era of basic research on gout had come to an end. The major players had left the field and NIH funding on the subject had dwindled to a trickle. Wyngaarden published his last research paper in 1977 and spent the years 1982 to 1989 as director of the National Institutes of Health. Kelley published his last papers on the genetics of gout in 1989, when he became dean of medicine at the University of Pennsylvania. Irving Fox, who did much of the basic research on fructose- and alcohol-induced hyperuricemia in Kelley’s lab, went to work in the biotechnology industry in the early 1990s. Only Edwin Seegmiller remained interested in the etiology of gout, and Seegmiller says that when he applied to the NIH for funding to study the relationship between fructose and gout, after elucidating the genetic connection with Radda in 1990, his grant proposals were rejected on the basis that he was too old and, as an emeritus professor, technically retired.(28) “In the 1950s and 1960s, we had the greatest clinical scientists in the world working on this disease,” says Kelley. “By the 1980s and 1990s, there was no one left.”
Meanwhile, the medical journals would occasionally run articles on the clinical management of the gout, but these would concentrate almost exclusively on drug therapy. Discussions of diet would be short, perhaps a few sentences, and confused about the science. On those occasions when the authors would suggest that gouty individuals might benefit from low-purine diets, they would invariably include “sugars” and “sweets” as among the recommended foods with low-purine contents.(29) In a few cases – a 1996 article in the New England Journal of Medicine, for instance (30)– the articles would also note that fructose consumption would raise uric acid levels, suggesting only that the authors had been unaware of the role of fructose in “sugars” and “sweets.” Even when the New England Journal published a report from Walter Willett and his Harvard colleagues in March 2004, this same kind of nutritional illiteracy manifested itself. Willett’s article had reported that men with gout seemed to eat more meat than healthy men. But Willett, who by this time was arguably the nation’s most influential nutritional epidemiologist, later explained that they had never considered sugar consumption in their analysis because neither he nor his collaborators had been aware of the hyperuricemic effect of fructose. Willett’s co-author, Gary Curhan, a nephrologist and gout specialist with a doctorate in epidemiology, said he might have once known that fructose raised uric acid levels, but it had slipped his mind. “My memory is not what it used to be,” he said. He also acknowledged, in any case, that he never knew sucrose was half fructose.
The addenda to this fructose-induced hyperuricemia story may be even more important. When the New England Journal of Medicine published Willett’s gout study, it ran an editorial to accompany it written by the University of Florida nephrologist Richard Johnson. Over the past decade, Johnson’s research has supported the hypothesis that elevating the uric acid concentration in the circulation also damages the blood vessels leading into the kidneys in such a way as to raise blood pressure directly, and so suggests that fructose consumption will raise blood pressure.
This is another potentially harmful effect of fructose that post-dates the official reports exonerating sugar in the diet. And it is yet another mechanism by which sugar and high fructose corn syrup could be a particularly unhealthy combination. The glucose in these sugars would raise insulin levels, which in turn would raise blood pressure by inhibiting the kidney’s secretion of sodium and by stimulating the sympathetic nervous system, as we discussed in an earlier chapter, and the fructose would do it independently by raising uric acid levels and so damaging the kidney directly. If this were the case, which has never been tested, it would potentially explain the common association of gout and hypertension and even of diabetes and hypertension.(31) Johnson is only now looking into this possibility, however. Unlike Willett and his colleagues, Johnson had long been aware of the ability of fructose to raise uric acid levels, and so was studying that phenomenon in his laboratory. But it was only in the summer of 2004, he explained, three months after his NEJM editorial was published, that he realized that sucrose was half fructose and that his research of the past years was even relevant to sugar.(32)
A decade later, Thomas Benedek described the epidemiology of gout in The Cambridge World History of Human Disease this way: “Worldwide the severity and prevalence of gout have changed paradoxically since the 1940s. In the highly developed countries, as a result of the advent of effective prophylactic drug therapy, the disease is now rarely disabling. Elsewhere, however, it has become more prevalent, predominantly as a result of `improved diets.’”
###
Footnotes and endnotes:
The economist and historian Ralph Davis estimates that the supply of sugar from the Caribbean into Britain rose from three or four thousand tons a year in the late fifteenth century to over two hundred thousand tons by the 1770s, or an increase of over fifty-fold. (davis r, the rise of the atlantic economies, cornell university press, 1973, p. 251, 255)
1 Kramer hm, curhan g, the association between gotu and nephrolithiasis: the national health and nutrition examination survey III. 1988-1994. Am J Kidney Dis 2002;40:37-42
2 Arromdee E, Michet CJ, Crowson CS, O’Fallon WM, Gabriel SE. Epidemiology of gout: is the incidence rising? J Rheumatol. 2002 Nov;29(11):2403-6.
2Interview with choi, sept 16, 2004
2Lawrence RC, Helmick CG, Arnett FC, Deyo RA, Felson DT, Giannini EH, Heyse SP, Hirsch R, Hochberg MC, Hunder GG, Liang MH, Pillemer SR, Steen VD, Wolfe F. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States.
2Arthritis Rheum. 1998 May;41(5):778-99.
3 gout, the patrician disease, p. 3
4
5 hydrick and fox, p. 748-749.
6 Duncan’s diseases of metabolism, p. 632.
7 Hydrick cr and fox ih, nutrition and gout, in present knowledge in nutrition, fifth edition, the nutrition foundation, Washington dc, 1984, p. 743
8 duncans diseases of metabolism, p. 638
9 Traut ef, rheumatic diseases, diagnosis and treatment, the C.V. Mosby Company, St. Louis, 1952 p. 303.
9benedek, in Cambridge history of diseases
9Trowel hc, a case of gout in a ruanda African, the east African medical journal, oct. 1947, p. 346-348
10 Beighton p et al, 1977, rheumatic disorders in th south African negro, part IV. Gout and hyperuricemia. South Af Med J. 51(26):969-72
11 Gout in the Maoris, B.S. Rose, Seminars in Arthritis and Rheumatism. Vol. 5, no. 2, (November) 1975, pg. 121-145.
12 duncan’s diseases of metabolism, 1947, p. 631
13 gertler mm, et al, erum uric acid in relation to age and physique in health andin coronary ehart disease, Ann Intern Med. 1951 Jun;34(6):1421-31. Reiser S, Uric Acid and Lactic Acid, in REISER S AND HALLFRISCH J, METABOLIC EFFECTS OF FRUCTOSE, crc press, boca raton fl, 1987 p. 113-134
13
14 duncan’s diseases of metabolism, p. 631
14 reaven gm, The Kidney: An Unwilling Accomplice in Syndrome X, Am J Kid Dis, Vol. 30, n0 6, December, 1997: pp. 928-931.
15 Facchini F et al, Relationship Between Resistance to Insulin-Mediated Glucose Uptake, Urinary Uric Acid Clearance, and Plasma Uric Acid Concentration, JAMA, December 4, 1991, vol. 266, no. 21, 3008-3011
16 Wyngaarden and Kelley p. ix
17 mintz
18 Sydney Mintz, Sweetness and Power, The Place of Sugar in Modern History, penguin books, ny 1985 p. 96.
19 mintz p. 64, 66
20 perheentupa j raivio k, fructose-induced hyperuricaemia, lancet, September 9, 1967, p.528531
21 emmerson bt, getting rid of gout
22 mayes pa, metabolism of fructose, ajcn, 1993
22hydrick c fox i, nutrition and gout, in modern reviews of nutrition
23 Seegmiller JE, Dixon RM, Kemp GJ, Angus PW, McAlindon TE, Dieppe P, Rajagopalan B, Radda GK. Fructose-induced aberration of metabolism in familial gout identified by 31P magnetic resonance spectroscopy.
23Proc Natl Acad Sci U S A. 1990 Nov;87(21):8326-30
24 peerheentupa ibid
25 hydrick and fox, p. 748-749.
26 Mayes pa, metabolism of fructose, ajcn 1993
27 Kelley interview
28 seegmiller interview
29 See for instance, fam ag, gout, diet and the insulin resistance syndrome, j. rheum. 2002;29, 1350-55
30 Emmerson BT. The management of gout.
30N Engl J Med. 1996 Feb 15;334(7):445-51
31 get citation from Richard Johnson articles on uric acid and hypertension.
link to Tims page

Recent Articles
-
Tony
May 08, 24 08:57 PM
Hi Peter, I am a physician, had my first Gout episode last week. I was surfing through the web and happened to come across your site. I am impressed by -
What is it about Cherries?
Aug 25, 21 03:59 AM
Allopurinol worked for me, but at the expense of adversely affecting my mood.I like cherries, but getting them every day of the year is a problem.I wondered -
ACV made my gout worse...
Apr 22, 20 07:19 AM
During my first gout attack, I was in so much pain that I desperately scoured the internet for alternative cures as the colchicine that I was prescribed
